
特集:神経変性疾患マーカー抗体
アルツハイマー病やハンチントン病研究に有用な抗体を紹介します。
神経変性疾患におけるタンパク質凝集
タンパク質のミスフォールディングや凝集は、様々な神経変性疾患に認められる神経変性疾患固有の特徴です。タンパク質凝集は、代表的な神経変性疾患であるアルツハイマー病からプリオンタンパク質が関与する希少疾患に至るまで神経変性疾患の顕著な特徴であり、衰弱性の病状と関連付けられます。凝集に関与するタンパク質は、各疾患によって異なります。ただし、以下に示すように凝集タンパク質が重複する疾患もあります。なおタンパク質凝集が原因となり神経変性疾患を発症するのか、神経変性疾患を罹患した結果タンパク質凝集が生じるのかについては明らかになっていません。特定の神経変性疾患に関する理解を深めることで、別の神経変性疾患の課題を解決する手掛かりを得られる可能性があります。タンパク質フォールディングの恒常性、そしてタンパク質凝集により恒常性を維持するプロセスが正常に働かなくなる機構の解明は、神経変性疾患の研究にとって非常に重要です。今後研究が進むことによって、神経変性疾患をターゲットとした有効性の高い治療法の開発につながることが期待されています。
タンパク質フォールディングの恒常性
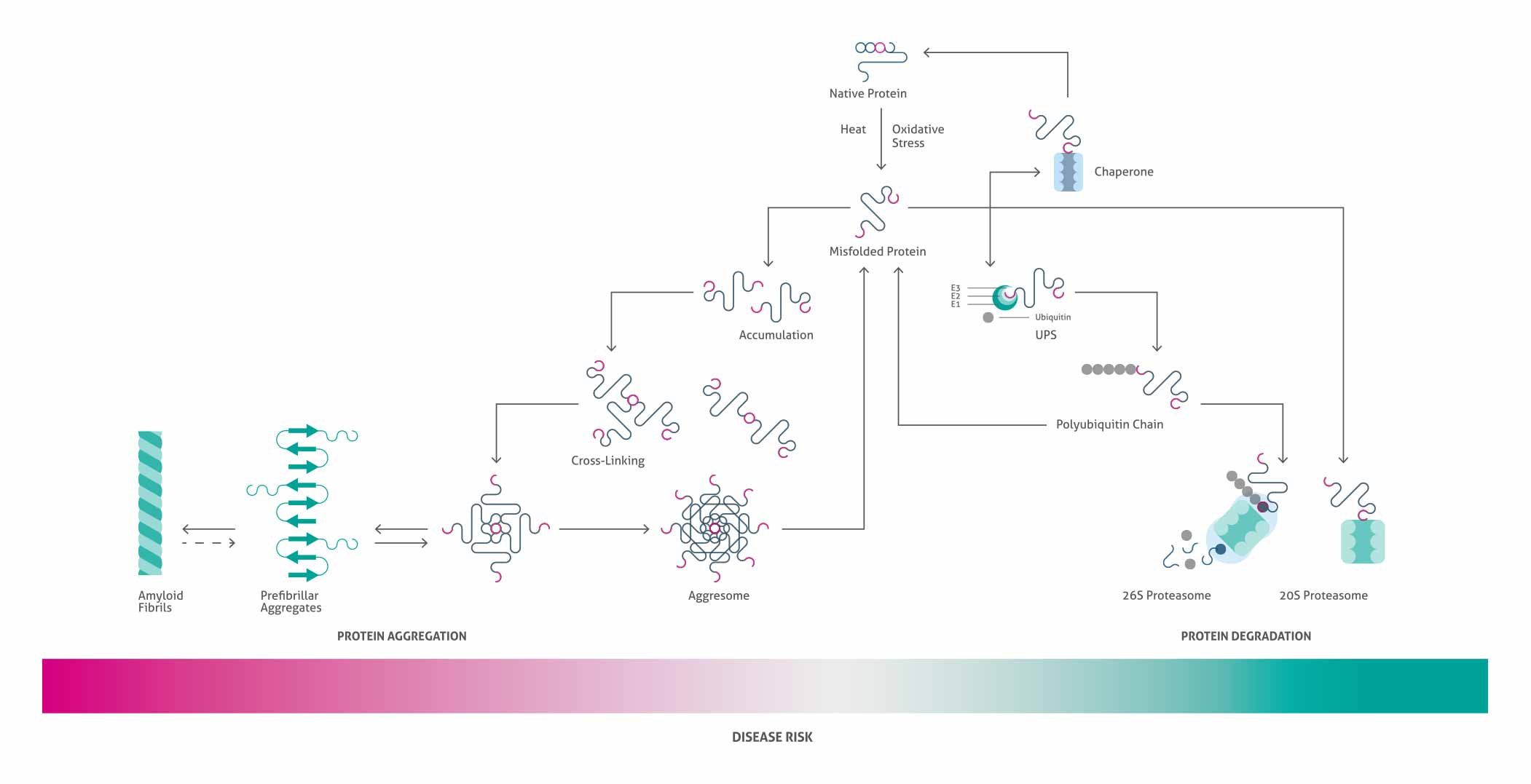
タンパク質のミスフォールディングは正常な細胞でも発生します。しかし、細胞にはミスフォールディングタンパク質に対処するためのメカニズムが備わっています。上の図に示されているように、ミスフォールディングタンパク質を正確に再度折りたたむ機構(例:タンパク質シャペロン)、あるいはタンパク質分解によりミスフォールディングタンパク質を分解する機構のいずれかが働きます。細胞の健全性を維持するために、このようなタンパク質の「トリアージ」システムは非常に重要です。タンパク質「トリアージ」の機構が機能不全に陥った場合や、ミスフォールディングタンパク質が細胞内に大量に発生してしまった場合、このバランスが崩れてしまい、タンパク質の「シャペロンによる折りたたみの促進」または「異常タンパク質の分解」から「異常タンパク質の蓄積」へとシフトします。線維状タンパク質の沈着を特徴とする疾患では、極端なタンパク質凝集が認められます。凝集タンパク質では、β-シートからなる異常な構造が不可逆的に架橋し、長い不溶性の線維状複合体を形成します。
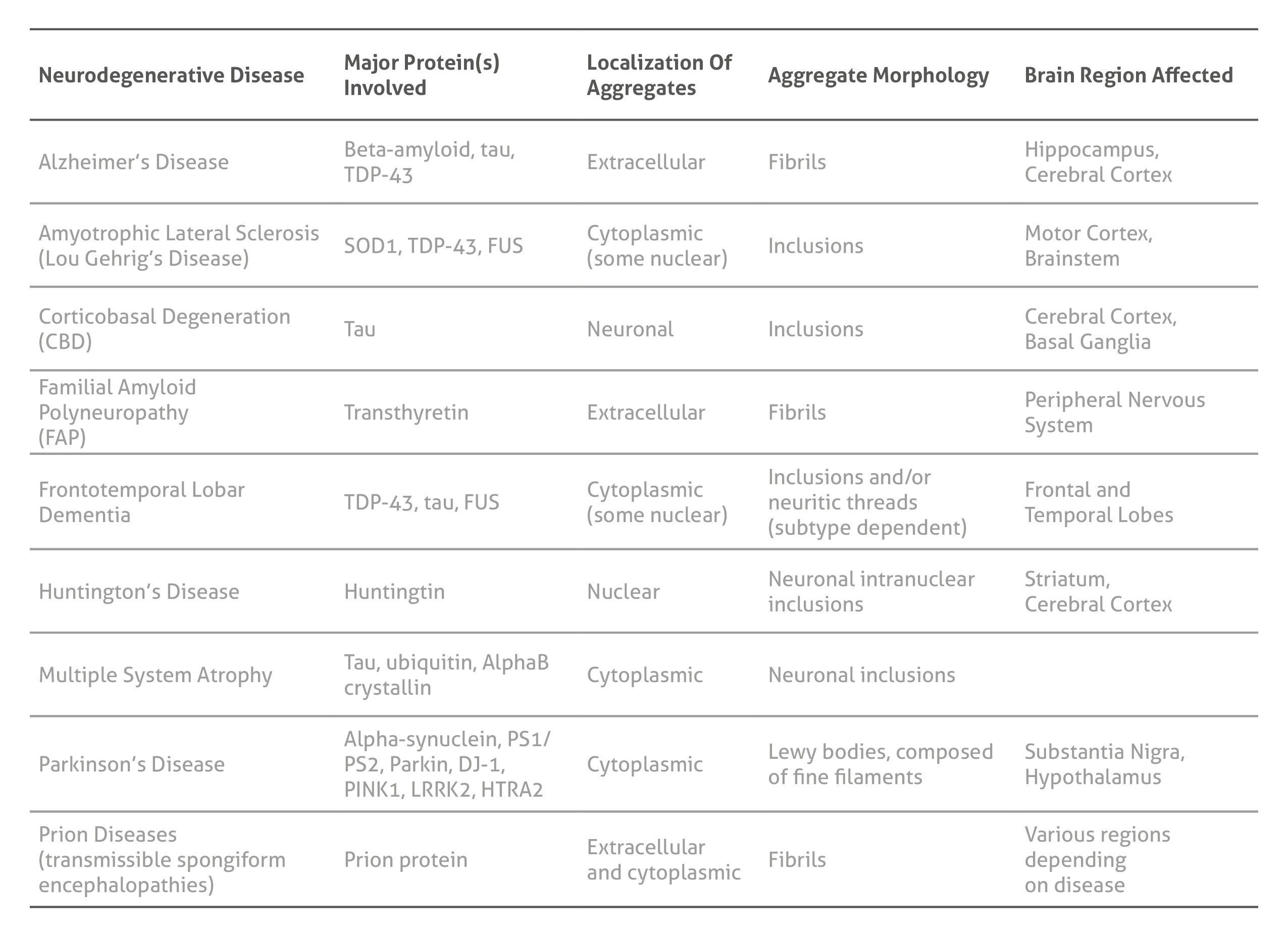
神経変性疾患に関連するタンパク質と病態
ダウンロード:神経関連疾患抗体カタログ[PDF](言語:英語) |
アルツハイマー病(Alzheimer's Disease)
アルツハイマー病(AD:Alzheimer's Disease)は、徐々に神経細胞が破壊し、重篤な認知機能低下を引き起こす慢性疾患です。最も一般的に知られる慢性疾患の形態がアルツハイマー型認知症です。アルツハイマー病は、老人斑(senile plaque)や神経原線維変化(NFT:neurofibrillary tangle)と関連します。アミロイドβ(Amyloid-beta)は、老人斑の主要な構成物質であり、細胞や臓器の機能に対して様々な病理学的影響を及ぼします。細胞外のアミロイドβオリゴマーは、細胞表面に存在するデスレセプター(Death receptor)の活性化を介して、カスパーゼの活性化を誘導すると考えられています。または、細胞内のアミロイドβがタウ(tau)タンパク質の過剰なリン酸化、ミトコンドリア機能の崩壊、カルシウムイオン恒常性の機能不全に伴うカルシウムイオンの増加を引き起こすことで、病態に影響すると考えられています。現在、遺伝子研究によりアルツハイマー病に関与する4種類の遺伝子が明らかになっています。4つの関連遺伝子は常染色体優性の遺伝子で、早期発症型家族性アルツハイマー病(Early-onset familial Alzheimer's disease)と関連する可能性が示唆されています。これらの4種類の遺伝子には、アミロイドβ前駆体タンパク質(APP:Amyloid precursor protein)、プレセニリン-1(PS1:Presenilin 1)、プレセニリン-2(PS2:Presenilin 2)、アポリポタンパク質E(APOE:apolipoprotein E)が含まれます。APP、PSタンパク質に関連する遺伝子変異はすべて、アミロイドβペプチドの中でも特に凝集性・神経毒性が高いアミロイドβ42の産生増加につながります。家族性アルツハイマー病に関連するPS1変異は、小胞体ストレス応答(UPR:unfolded protein response)を下方制御し、小胞体(ER)ストレスに対する脆弱性に寄与する可能性を示唆されています。
ダウンロード: アルツハイマー病シグナル伝達パスウェイポスター[PDF] |
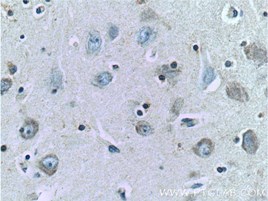
| TAU | TDP-43 |
 |
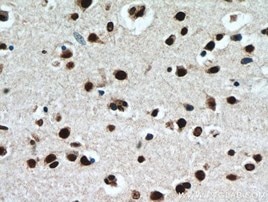 |
| TAU抗体(カタログ番号:10274-1-AP、希釈倍率1:200)を使用したパラフィン包埋ヒト脳組織スライドの免疫組織化学染色(40X)。 | TDP-43抗体(カタログ番号:10782-2-AP、希釈倍率1:50)を使用したパラフィン包埋ヒト脳の免疫組織化学染色(40X) |

パーキンソン病(Parkinson's Disease)
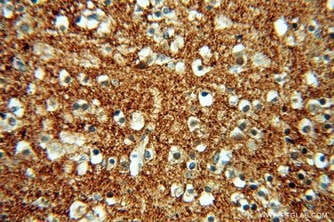
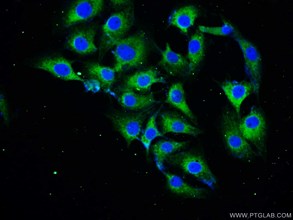
パーキンソン病(PD:Parkinson's Disease)は、中枢神経系の運動系に影響を及ぼす、時間をかけて進行する中枢神経系疾患です。シヌクレイン(synuclein)遺伝子の変異は、いくつかの家族性のパーキンソン病発症の原因となります。パーキンソン病の基本的な病態は、レビー小体(神経細胞に認められる封入体構造)におけるシヌクレインタンパク質の凝集です。
 |
 |
|
a-synuclein抗体(カタログ番号:10842-1-AP、希釈倍率1:50)を使用したパラフィン包埋ヒト脳の免疫組織化学染色(40X)。 |
a-synuclein抗体(カタログ番号:10842-1-AP、希釈倍率1:25)、Alexa Fluor®488 AffiniPure Goat Anti-Rabbit IgG(H+L)抗体を使用したSH-SY5Y細胞の免疫蛍光染色。 |
ハンチントン病(Huntington's Disease)
ハンチントン病(HD:Huntington's Disease)は、舞踏運動等の進行性の不随意運動、認知障害、行動障害等の精神症状等を特徴とする常染色体優性の進行性神経変性疾患です。ハンチントン病は、ハンチンチン(Huntingtin)タンパク質をコードするHTT遺伝子の変異を原因とする疾患です。
