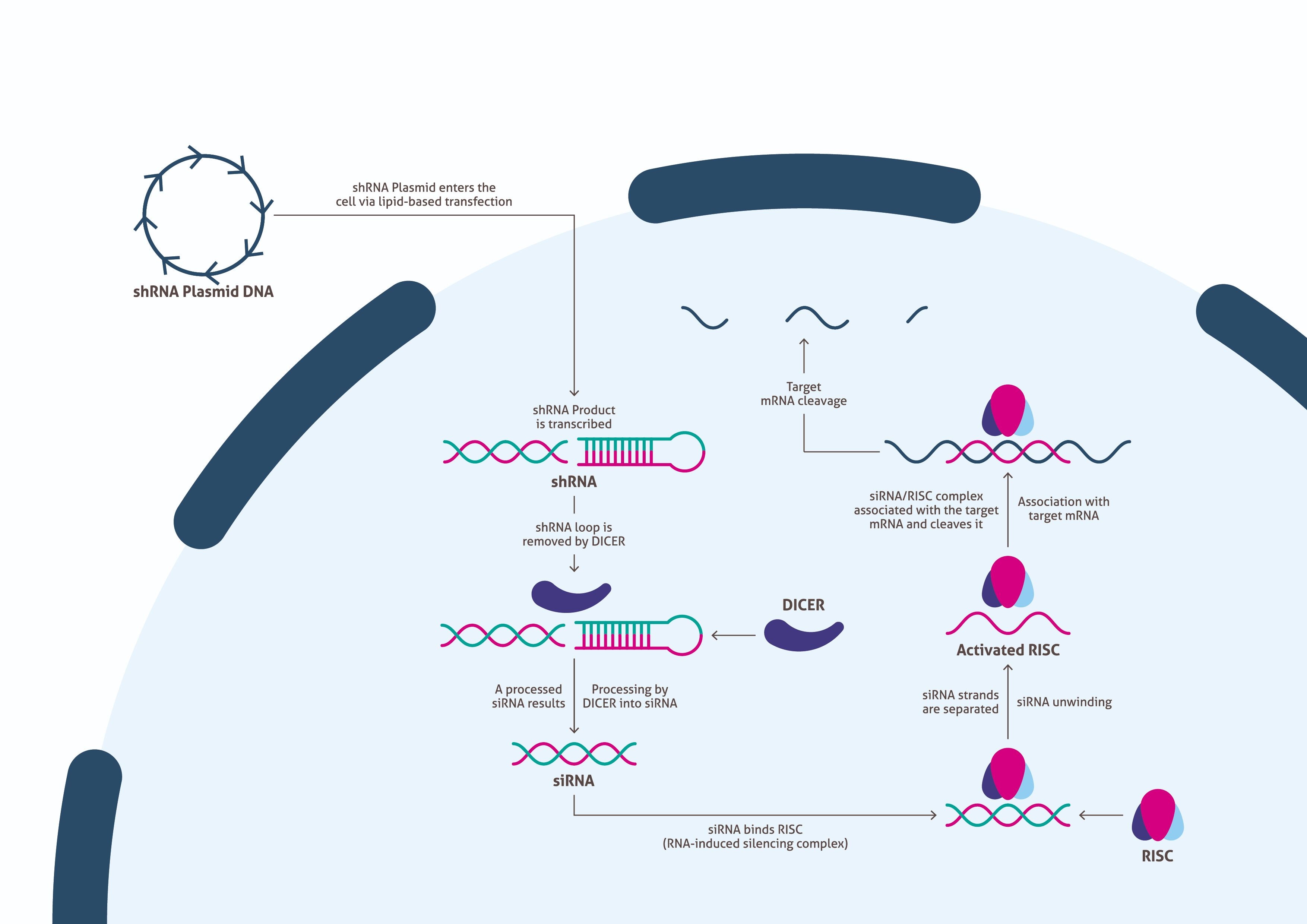
ベクターを作製し、トランスフェクション用に十分な量を確保したのであれば、ベクターを細胞に送達するための適切なトランスフェクション法を決定しなければならない。トランスフェクションが成功すると、細胞は外部から導入されたDNAを転写して上述したshRNAを産生する(図2)。その後、Dicerがループ配列を除去することで、shRNAはsiRNAにプロセシングされる。プロセシングを受けたsiRNAはRISC(RNA-induced silencing complex)に結合し、RISCは二本鎖RNAを分離させて活性化する。RISCにはsiRNAの片方のRNA鎖が残り、ターゲットmRNAと相補的に結合してターゲットmRNAを切断する。この機構により、ターゲットmRNAにコードされたタンパク質の生成が抑制される。研究者は、このRNAi実験を実施するにあたって、ウェスタンブロットに使用するための十分な量のサンプルを得るために細胞を培養すると同時に、RNAiの生物学的プロセスが完了するまでの時間を確保する必要がある。
ターゲットタンパク質が細胞生存に必須のタンパク質である場合、細胞死は実験を頓挫させる主要な原因となる。幸いなことに、タンパク質合成が抑制されても、完全に消失していなければ、ほとんどの細胞は正常に増殖することが可能である。必須タンパク質をターゲットとする場合は、ノックダウン法の方が発現抑制の微調整が可能であり、遺伝子編集によってターゲットの転写を全て消失させるノックアウト法による検証試験よりも、好ましい代替法と考えられる。
試験および評価(Test and evaluate)
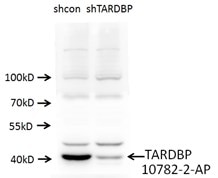
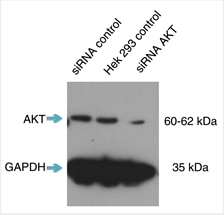
ウェスタンブロットを実施した際、shRNA発現ベクターを導入しなかった細胞では強いシグナルが認められ、shRNA発現ベクターのトランスフェクションを実施した細胞では弱いシグナルが認められた場合、それは抗体がターゲット特異的でありノックダウン実験が成功したことを意味している。何らかの非特異的バンドが認められる場合は、抗体自体が非特異的であることを示している可能性があるため、抗体の品質に疑念が生じる。また、観察されるバンドはウェスタンブロットメンブレンのすべてのレーンにおいて整合しているはずである。そうでない場合は、何らかの原因によって実験が失敗している可能性があり、プロトコールおよび実験計画を再検討する必要がある。
抗体検証プロトコールのネガティブコントロールの重要性を過小評価すべきではないと考える一方、このような検証方法は広く一般的に利用可能な方法ではなく、個別のラボで実施するには時間とリソースを消費する手法であることも十分に理解できる。その際、次善の選択肢としては、ポジティブコントロールのみを採用して綿密にウェスタンブロットによる検証を実施する手法が挙げられる。ただし、対象となる抗体製品に対して十分な数のデータが揃っていること、つまり使用した抗体のメーカー名や品番が明記されている研究論文が発表されていることが条件となる。我々は、抗体検証における新たなスタンダードとして、siRNAノックダウン等のネガティブコントロールの展開と利用を奨励したい。そうすることで、再現性の高い研究成果をもたらし、科学の発展を加速させるはずである。
参考文献