
Western Blot Troubleshooting: Why Does The Observed Protein Molecular Weight (MW) Differ From The Calculated One?
リンク
- Western Blot Protocol
- How To Optimize Your Western Blot
- SDS-PAGE Gel Recipes
- How To Optimize Your Results With Low MW Proteins
- Tricine Gel Recipe For Low Molecular Weight Proteins
- Choosing The Right Lysis Buffer
- Choosing The Right Western Blot Detection Method
- Western Blot Troubleshooting: Why Does The Observed Protein Molecular Weight (MW) Differ From The Calculated One?
- Western Blot Troubleshooting: High Background
- Western Blot Troubleshooting: Weak/No Signal & Other
- Western Blot ppt
- Western Blot Video Protocol
Western blotting and Molecular Weight (MW)
The first WB step is sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by protein transfer on a membrane and subsequent detection with specific antibodies. Because the SDS-PAGE is conducted in denaturing conditions, proteins migrate according to their molecular weights irrespective of their secondary/tertiary structure, charge or protein–protein interactions. This means that smaller proteins migrate faster than larger ones.
The predicted molecular weight (MW) of the protein is the sum of the molecular weights of all protein amino acids. It is easy to calculate, e.g., using the free online ExPASy tool. Often the calculated MW is different from that observed on the WB. Here we try to summarize the most common reasons for why this may occur (Figure 1).
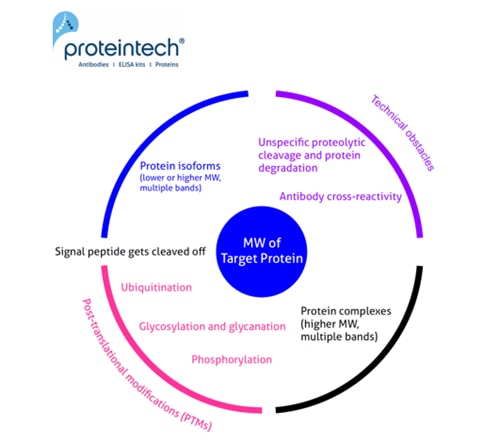
Unusual or unexpected size of WB bands
1. Signal peptide (and a pro-peptide) gets cleaved off
Many proteins that undergo transport through the secretory pathway have signal peptides of 15–35 aa. length located predominantly at their N-termini. They are often cleaved by various proteases during their subcellular transport. This results in the mature protein running at a lower than predicted molecular weight. The presence of signal peptides can be predicted by various online tools or based on previously published data. They are usually well annotated in protein databases, e.g., UniProt. Additionally, a subset of proteins has pro-peptides – protein domains that are present in protein precursors. Protein precursors need to be processed by proteases in order to engender a functional product (without pro-peptide).
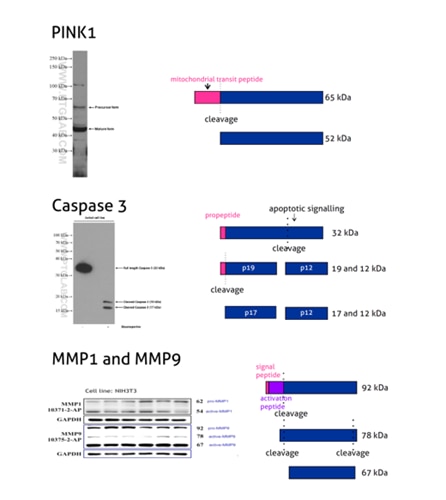
Figure 2: PINK1 (23274-1-AP) is a mitochondrial serine/threonine-protein kinase that protects cells from stress-induced mitochondrial dysfunction. The precursor of PINK1 (65 kDa) is synthesized in the cytosol and is imported into the outer membrane of mitochondria. PINK1 is further transferred into the inner membrane where it is cleaved into a 52 kDa mature form.
Caspases, a family of endoproteases, are critical players in cell regulatory networks controlling inflammation and cell death. Caspase 3 (19677-1-AP) exists as an inactive proenzyme form of 32 kDa (p32), which upon apoptotic signaling gets cleaved into two active subunits (p19/17 and p12) that assemble into a functional tetrameric enzyme (PMID: 7596430).
Proteins of the matrix metalloproteinase (MMP) family are involved in the breakdown of the extracellular matrix in many physiological processes, including embryonic development, reproduction, tissue remodeling, and disease processes such as arthritis or metastasis. Most MMPs are secreted as inactive proproteins that are activated when cleaved by extracellular proteinases. The inactive pro-MMP9 (10375-2-AP) is 92 kDa. It gets sequentially cleaved by MMP3 into a processed form of 68 kDa through an intermediate form – 78/82 kDa (PMID: 1371271). Besides, MMP9 can also exist as a dimer of 180 kDa (PMID: 7492685).
2. Posttranslational modifications (PTMs)
a) Glycosylation and glycanation
The majority of proteins that are synthetized on ribosomes associated with the endoplasmic reticulum undergo glycosylation. That means a covalent attachment of sugar moieties is added to the polypeptide chain. The two most common types of glycosylation in Eukaryotes are N-linked glycosylation – to asparagine, and O-linked glycosylation – to serine and threonine. Extensive glycosylation adds additional molecular weight, not included in the original protein sequence, which makes proteins migrate slower.
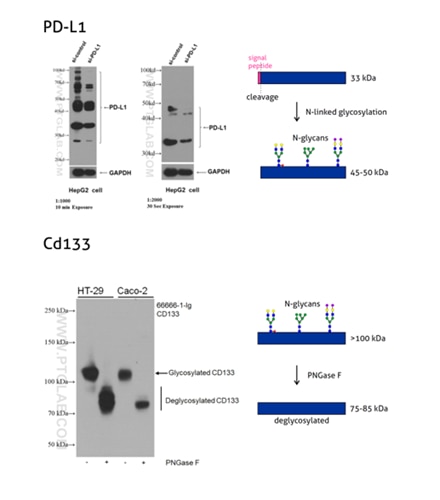
Figure 3: Programmed cell death ligand 1 (PD-L1, CD274, or B7-H1) (66248-1-Ig) is a type I transmembrane protein, acting as a key regulator of the adaptive immune response. Full-length PD-L1 MW is 33 kDa. The signal peptide is cleaved off during protein transport to the plasma membrane and the protein is heavily N-glycosylated with an apparent molecular weight of 45–70 kDa with the major glycosylated form of 45–50 kDa (PMID: 27572267).
Please note: An enzymatic deglycosylation is a commonly used experimental technique to verify whether a studied protein is glycosylated. Prior to WB, the protein sample is incubated with an enzyme that is able to remove parts or full glycan chains. WB protein species from the digested sample are then compared with the undigested sample, and any observed shift in molecular weight indicates protein glycosylation. One commonly used enzyme is PNGase F; it removes N-linked glycans by cleaving the bond between the innermost N-Acetylglucosamine of the glycan chain and the asparagine residue.
CD133, also known as PROM1 (prominin-1) (18470-1-AP), is a transmembrane glycoprotein with an NH2-terminal extracellular domain, five transmembrane loops, and a cytoplasmic tail. CD133 is a highly glycosylated protein with an apparent molecular weight of 115-120 kDa. After lysates treatment with PNGase F, CD133 shifts to a protein with an MW of 75–85 kDa. That corresponds to the calculated molecular weight of deglycosylated CD133 (PMID: 23150174).
Proteoglycans are a special case group of glycoproteins. They are extracellular matrix proteins with long unbranched glycosaminoglycan chains covalently attached to the amino peptide chain core. Usually, the molecular weight of the sugar group is even larger than the protein component.
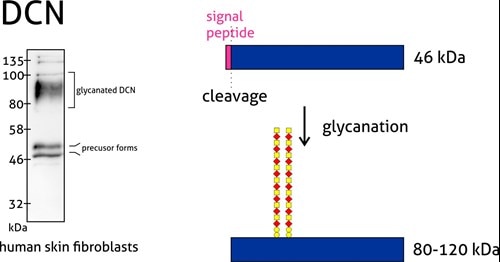
Figure 4: Decorin (14667-1-AP) is a member of the small leucine-rich proteoglycan family of proteins. Decorin precursor forms a range of 43–47 kDa MW. It contains a cleavable N-terminal peptide signal and can also be glycosylated. The attachment of glycosaminoglycans (chondroitin sulfate or dermatan sulfate) to decorin occurs in the Golgi apparatus prior to secretion of the mature glycanted form from cells.
b) Phosphorylation
One of the most common posttranslational modifications is protein phosphorylation. It takes place on serine, threonine, and tyrosine residues. Phosphorylation regulates protein function, its enzymatic activity, protein–protein interactions, and protein localization. Phosphorylation is catalyzed by phosphatases and can be reversible – phosphorylated proteins can be dephosphorylated by protein dephosphatases. The addition of a single phosphoryl group adds +/- 1 kDa to the MW, which is often beyond the resolution of the standard SDS-PAGE. However, multiple phosphorylation sites can lead to more prominent MW changes.
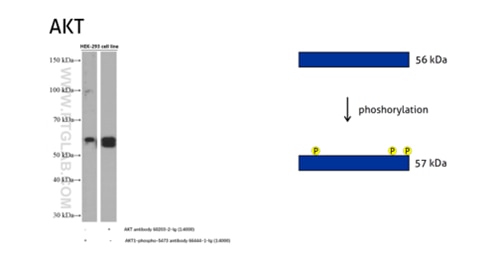
Figure 5: The serine/threonine-protein kinase AKT plays a role in many cellular processes. Survival factors can suppress apoptosis in a transcription-independent manner by activating the serine/threonine kinase AKT1, which then phosphorylates and inactivates components of the apoptotic machinery. (60203-2-Ig detects all the AKT members with or without phosphorylation, 66444-1-Ig detects the phospho-Ser473 of AKT1 and phospho-S474 of AKT2/phospho-Ser472 of AKT3.)
c) Ubiquitination
Protein ubiquitination means a covalent ubiquitin is added to lysine, cysteine, serine, threonine, or directly to the protein N-terminus. Ubiquitin is a small (+/-8.6 kDa) protein expressed across almost all tissue types. Ubiquitination is an enzymatic reaction catalyzed by a three-enzyme cascade (E1, E2, and E3). That provides substrate specificity and activation, conjugation, and ligation steps. Proteins can be monoubiquitinated (with one ubiquitin molecule) or polyubiquitinated. Polyubiquitination takes place when additional ubiquitin molecules are added to the initial ubiquitin molecule. Ubiquitination via the proteome can mark proteins for degradation. It is also important for cellular signaling, the internalization of membrane proteins , and the development and regulation of transcription. Ubiquitin can be removed from proteins by deubiquitinating enzymes, which lowers their MW.
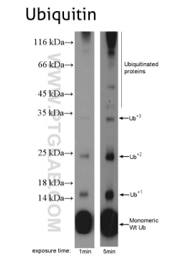
Figure 6: Ubiquitin B (UBB) (10201-2-AP), a member of the ubiquitin family, is required for ATP-dependent, non-lysosomal intracellular protein degradation of abnormal proteins and normal proteins with a rapid turnover. This gene consists of three direct repeats of the ubiquitin coding sequence with no spacer sequence.
3. Protein complexes
WB SDS-PAGE is performed in reducing conditions. That means that the majority of the protein complexes composed of proteins linked via non-covalent bonds disassociates during sample preparation and electrophoresis, and (individual) proteins run as monomers. However, some proteins remain partially or fully present in homo- or hetero-meric complexes, even in the presence of SDS and β-Mercaptoethanol. In this case, their observed molecular weight can be substantially higher than the predicted, calculated monomeric form. Some proteins, especially transmembrane proteins and proteins with hydrophobic domains, can aggregate during cell lysis as they are released from their native protein complexes and lipid membranes. These aggregates have high molecular weights and may not represent interactions that occur in their native states.
Please note: 20% β-Mercaptoethanol (or 100 mM DTT) for the 4X SDS sample buffer might help to remove unspecific bands due to dissociation of the protein complex.
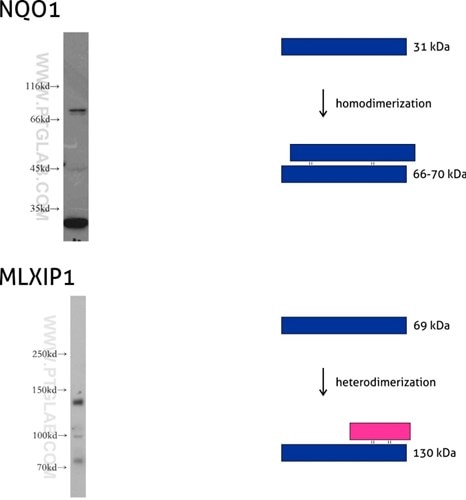
Figure 7: NQO1 (11451-1-AP) is an enzyme that serves as a quinone reductase together with conjugation reactions of the hydroquinones involved in detoxification pathways as well as in biosynthetic processes such as the vitamin K-dependent gamma-carboxylation of glutamate residues in prothrombin synthesis. NQO1 has three isoforms of 26, 27, and 31 kDa MW, and the formation of homodimers (66-70 kDa) is needed for its enzymatic activity.
Mlx-interacting protein (MLXIP, also known as MONDOA) (13614-1-AP) acts as a transcription factor by forming a heterodimer with MLX protein. This complex binds to and activates transcription from CACGTG E boxes, playing a role in the transcriptional activation of the glycolytic target and glucose-responsive gene regulation. MLXIP has three isoforms: 110, 57, and 69 kDa, and the molecular weight of the MLXIP-MLX heterodimer is 130 kDa.
4. Protein isoforms
Many proteins encoded by a single gene exist in more than one sequence variant – called protein isoforms. They arise from alternative splicing during mRNA maturation. Selected exons and introns can be included/excluded from the final mRNA product. Included additional protein-coding sequences can reflect in higher MW protein products. On the other hand, the addition of sequences can introduce alternative (premature) stop codons, leading to proteins of lower molecular weights. Some proteins have multiple translation start sites, which gives rise to isoforms with different N-termini. Protein isoforms can have a different half-life and subcellular localization. They can interact with different subsets of proteins, form distinctive protein complexes as well as have different, even opposite, functions.
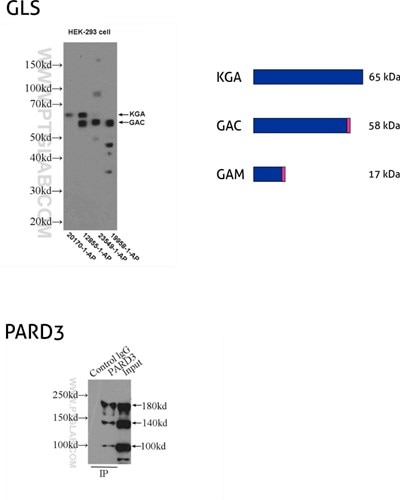
Figure 8: GLS, also known as GLS1 and KIAA0838, belongs to the glutaminase family. Three isoforms of GLS, named KGA, GAM, and GAC, vary in their MW and tissue expression patterns (PMID: 11015561). KGA, kidney-type glutaminase, has an MW of 65 kDa. GAC, glutaminase C, is 58 kDa, being a product of gene splicing that results in loss of the C-terminal domain that is present in KGA. GAM is the shortest isoform with no catalytic activity and comes into being from the inclusion of intron 2 and premature stop codon. (12855-1-AP detects KGA and GAC isoforms, 20170-1-AP is specific to KGA, 23549-1-AP is specific to all three (KGA, GAM, GAC) isoforms of GLS, and 19958-1-AP is specific to GAC.)
PARD3 (also known as ASIP, Par3, or Bazooka) is one of the PARD family proteins involved in asymmetric cell division and polarized growth. PARD3 has multiple splice isoforms with three main ones: 100 kDa, 150 kDa, and 180 kDa. (11085-1-AP recognizes all three main isoforms of PARD3.)
5. Technical obstacles
a) Antibody cross-reactivity
It is possible for the selected antibody to recognize not only its target protein but also cross-react nonspecifically with other proteins in the analyzed sample. To avoid this issue we might set up an appropriate controls panel and protocol optimization.
Suggested controls:
- Options for positive controls:
- a purified target protein
- a lysate from a cell line known to express the target protein
- a lysate from a cell line overexpressing the target protein.
- Negative controls:
- lysates from cell lines with lower expression of the target protein
- lysates from cell lines with the target protein knocked down (e.g., by siRNA or shRNA) or knocked out (e.g., by CRISPR).
Experimental optimization:
- Extraction buffers (e.g., RIPA buffer)
- Blocking buffers (e.g., 5% skimmed milk, casein, or BSA)
- Incubation and washing times (e.g., overnight at 4C or 1.5h at RT)
- Secondary antibodies used for detection (e.g., dilution factor)
- Membranes types (nitrocellulose vs. PVDF).
b) Unspecific proteolytic cleavage and protein degradation
Proteins can undergo unspecific proteolytic digestion if the protein sample is not correctly handled. Proteases released during cell lysis or tissue extraction can cause protein fragmentation, resulting in fragments of lower molecular weights. Some proteins are more susceptible to degradation than others. The choice of cell/tissue lysis buffers and lysis conditions, along with supplementation with protease inhibitors, are vital elements for efficient protein extraction.
Summary table
|
Observed molecular weight |
Potential causes |
|
Higher than expected |
1. Posttranslational modifications. 2. Antibody is detecting a protein isoform with a longer sequence. 3. Protein complexes. |
|
Lower than expected |
1. Cleavage of signal peptide. 2. Antibody is detecting a protein isoform with a shorter sequence. 3. Unspecific protein cleavage. |
|
More than one band observed |
1. Protein isoforms. 2. One protein product but with different posttranslational modifications. 3. Antibody is detecting protein with and without pro-peptide. 4. Protein complexes. 5. Antibody cross-reactivity, potentially due to homology of the immunogen sequence. |
|
No bands observed |
1. Protein cleavage. 2. Protein degradation. - Make sure to include appropriate controls. - Further protocol optimization is possibly needed. |