
質量分析の要点解説/必須事項
質量分析法におけるサンプル調製、サンプル精製およびデータ分析の概要を説明します。
質量分析法(MS:mass spectrometry, mass spec)は、高分子を解析する強力な手法です。
質量分析法は、単純な組成のサンプルや複雑な組成のサンプルをイオン化して、得られる質量(m)と電荷(z)を基にした分析手法です。
分析機器と解析ソフトウェアの両方が急速に進歩した恩恵により、21世紀の間に様々なアプリケーションの適用範囲と汎用性は著しく拡大しています。質量分析法は、依然として「単純な」化合物やペプチドの解析と同定に頻繁に使用されていますが、最新のアプローチでは大規模なデータセットを対象とした解析に一層注力しています。
質量分析法は、3つのステップに分かれます(図1)。
1. サンプル調製(Sample preparation)
2. サンプル精製(Sample purification)
3. データ解析(Data analysis)
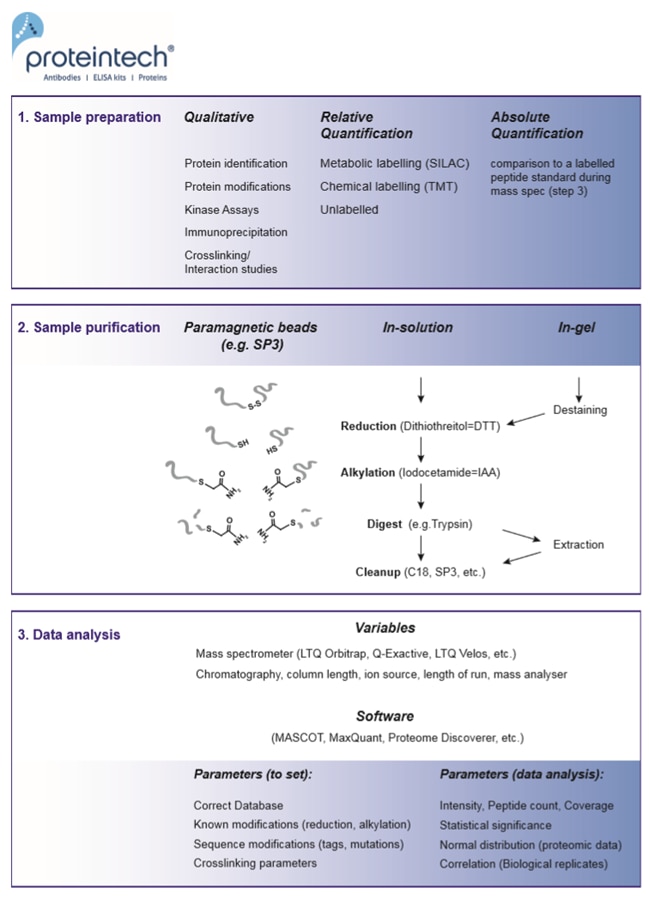
図1. 質量分析の3ステップ(1. サンプル調製、2. サンプル精製、3. データ解析)
1. サンプル調製
タンパク質存在量
質量分析法は、フェムトグラム(fg)レベルのタンパク質を検出することができる、最も感度の高いタンパク質検出法の1つです。
しかし、こうした性能を発揮できない場合があり、その際はモニタリングやバリデーションを実施する必要があります。細胞内タンパク質濃度が低い場合、タンパク質が分解されやすい場合、タンパク質が特定の細胞小器官でのみ発現している場合等において、存在量の多い他の細胞質内タンパク質が、複雑なサンプル中で測定時に目的タンパク質のシグナルをマスクする可能性があります。
サンプルの質量分析を実施する前に起こり得る問題を予測するため、SDS-PAGEや可能であればウェスタンブロット解析を実施し、全体を通して実験のモニタリングを行うと良いでしょう。
プロテインテックのアプリケーションサポート:ウェスタンブロット解析プロトコール(Western blot protocol)
低濃度のタンパク質を濃縮するためには、免疫沈降法や細胞分画法を検討することも有効な手段です。細胞分画を実施する場合は、ウェスタンブロットで細胞小器官マーカーを並行して確認し、各細胞器官が適切に分離していることを確認します。
プロテインテックのアプリケーションサポート:免疫沈降法(Immunoprecipitation)
コントロール
実験方法によって、実施する必要のあるコントロールは異なります。
定性的実験:
免疫沈降法やキナーゼアッセイ等が挙げられます。この場合、例えば「抗体を添加しないで実施する」、「タグ標識されていないサンプルを使用する」、「キナーゼを添加しないで実施する」、「野生型と変異型を比較する」といった、通常の生物学的コントロールを実施します。
相対定量:
相対定量には、代謝標識(SILAC: Stable Isotope Labelling by Amino acids in Cell culture、細胞培養物中のアミノ酸を使用した安定同位体標識法)や、化学標識(TMT: Tandem Mass Tag、タンデムマスタグ分析法)等があります。これらの方法の概要については、図2を参照してください。最大3種類のサンプル(SILAC)または10種類のサンプル(TMT)間のペプチド量の違いをモニタリングすることができます。複数の同位体標識アミノ酸を使用することは、得られたデータセットを検証するために非常に重要となります。すなわち、標識を入れ替えたサンプルを用いて、生物学的な反復解析を実施することで、実験手法の誤差を排除するようにします。また、最も低価格で、精度の低い手法として、ラベルフリー(非標識)定量法もあります。この方法では2回の別々の測定で得られたスペクトルをAUC(曲線下面積)またはスペクトルカウントで比較します。
絶対定量(AQUA):
既知濃度の標識合成ペプチドを、質量分析するサンプル中に「スパイクイン」します。標識ペプチドを内部標準として添加して目的物質を定量するため、この手法は非常に精度の高い手法です。しかし、標識ペプチドを合成する必要があるため、コストは比較的高くなります。
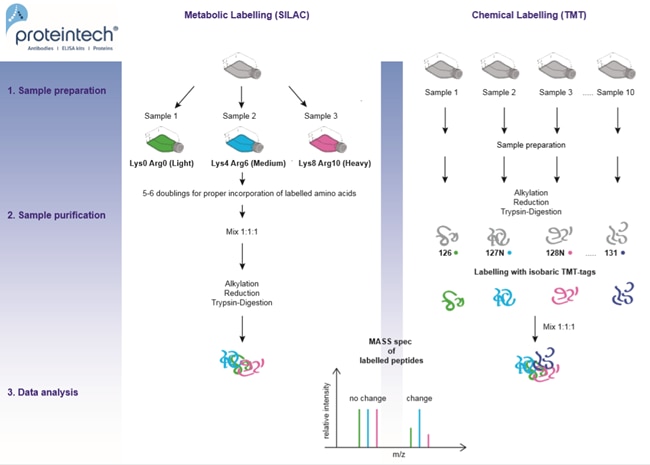
図2. 相対的定量法:代謝標識(SILAC)と化学標識(TMT)
タンパク質の安定性とその修飾
サンプル中にプロテアーゼ(またはホスファターゼ)が存在する可能性がある場合は、必ず十分量の阻害剤を添加してください。
実験中は、タンパク質分解活性を最小限に抑えるために、できる限り多くのステップを低温(4℃)で実施することを推奨します。なお、プロテアーゼ阻害剤は、トリプシン消化(またはその他の使用プロテアーゼ)を阻害するため、質量分析のサンプル調製の前に、サンプル溶液から除去する必要があります。
汚染物質
例えば、ケラチンの混入(コンタミネーション)を避けるために、サンプルが触れる可能性のある表面、容器、チップ、溶液を清潔にしてください。ケラチンは特に存在量が多く、シグナルをマスクします。
また、ポリマーの中でも、特にPEG(ポリエチレングリコール)は汚染の原因となります。PEGは、ガラス製品の洗浄に使用される洗剤に由来するか、またはオートクレーブ滅菌中にプラスチック容器やチップから溶出します。
すべての使用溶液にはHPLCグレードの水を使用し、オートクレーブ滅菌されていないフィルターチップ、目的別に用意したピペット、タンパク質が結合しにくい(タンパク質低吸着)チューブを使用すると良いでしょう。時には、アフィニティーカラムの素材からもポリマー汚染が生じます。その場合、追加の洗浄ステップとして、トリプシン処理や抽出を実施する前にサンプルをゲルに通す操作を検討してください。プレキャストゲルは、ケラチン汚染のリスクが少ないためおすすめです。自作ゲルの場合は、HPLCグレードの水を使用し、エタノールですべての器材を徹底的に洗浄します。
2. サンプル精製
酵素消化
還元およびアルキル化後、タンパク質を適切なサイズのペプチドに消化する必要があります。検出に最適なペプチド長は8~15アミノ酸です(1)。
最も一般的に使用される消化酵素は、トリプシンです。消化後のペプチド長は、無料で利用できるオンラインソフトウェアPeptideMass(https://web.expasy.org/peptide_mass/)を使用して予測することができます。トリプシン認識部位が十分にないか、または多すぎる場合は、他のプロテアーゼを使用して、より適切な鎖長のペプチドを生成します。
ゲル内または溶液中での消化
タンパク質は、溶液中で直接消化することも、ゲルにロードした後にゲル内消化を実施してペプチドを抽出することも可能です。
| 溶液中処理 | |
| 利点 | サンプルからの損失がない |
| 欠点 | バッファーはトリプシン消化処理と互換性のあるものでなければならない |
| ゲル中処理 | |
| 利点 | ポリマーのような汚染物質のコンタミネーションを避ける追加的洗浄ステップとなり、また泳動レーンを別々の画分に切断することで、分子量に従いタンパク質を更に分離できる |
| 欠点 | サンプルの損失がある |
クリーンアップ
汚染物質、塩類、バッファーを除去するため、クリーンアップは、サンプルを質量分析で測定する前の、非常に重要な最後のステップとなります。疎水性ペプチドを結合するC18カラム(他には逆相樹脂のカラムがあります)が最も一般的に使用されており、塩類や親水性の汚染物質をウォッシュアウトします。
次に、ペプチドを有機溶媒で溶出します。SP3(Single-Pot Solid-Phase-enhanced Sample Preparation)のような最新の方法では、タンパク質を磁気ビーズに結合させて、洗浄と消化を直接行うことができるため、より厳密な洗浄が可能となり、サンプルからの損失を最小限に抑え、再現性が高くなります(2)。
3. データ解析
データベース
- UniProt(https://www.uniprot.org)から自分の実験や生物種に適したタンパク質データベースを選択し、その最新の配列情報を質量分析ソフトウェアにアップロードします。
- エピトープタグが含まれているタンパク質の場合は、タグ配列を含めたタンパク質の配列をデータ解析用のデータベースで使用してください。
可逆性架橋剤(可逆性クロスリンカー)
- サンプル精製前、架橋剤(クロスリンカー)は化学的に切断され、相互作用するタンパク質は追跡可能な修飾を受けた状態になります。この修飾をデータベースに含めます。
非可逆性架橋剤(非可逆性クロスリンカー)
- 架橋ペプチドは、異なるタンパク質に由来することがあるため、データの解析には専用のソフトウェアとガイダンスを使用します。架橋ペプチドの存在はデータ解析をさらに複雑にします。
再現性
- プロテオームのデータセット全体を解析する場合は、1回の測定で検出されるすべてのペプチドのスペクトルが、ガウス分布(正規分布)に従うことを確認します。
- 異なる生物学的反復(レプリケート)から得られたデータセットは、互いに一致していなければなりません。そうでない場合、結果は無効となります。
- ペプチドの測定結果/同定結果は統計的に有意でなければなりません。p値(ソフトウェアによっては、Q値またはスコア)は<0.05とします。