
ヒントとコツ | ChIP実験を成功させるための6つのヒント
ChIP実験で良好な結果を得るための、6つのポイントを解説します。
クロマチン免疫沈降(ChIP)の概要
|
クロマチン構造が遺伝子発現の調節で重要な役割を果たしていることを示すエビデンスは増加しており、クロマチン構造解析の重要性はますます高まっています。ChIPアッセイの実施によって、遺伝子調節の構造的側面と機能的側面を両方同時に研究することができます。以下に、ChIP実験の最適化に役立つ最も重要なヒントをまとめました。
ChIPプロトコール詳細はこちら[PDF] |
ChIPフローチャート |
IPプロトコール詳細はこちら[PDF] |
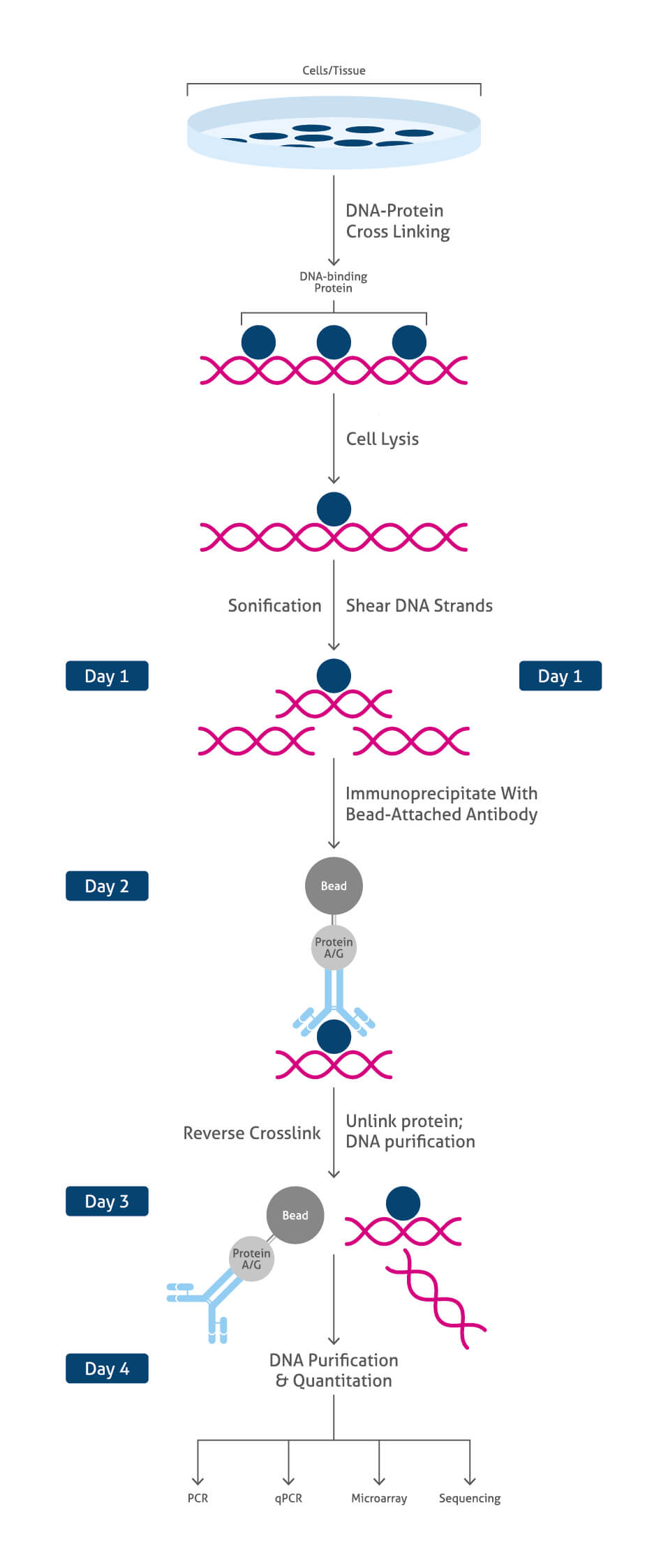
ワークフローの概要
- 細胞/組織を単離する。
- 目的タンパク質がクロマチンと結合している状態でDNA‐タンパク質複合体を架橋(クロスリンク)して固定する。
この段階で操作を中断することが可能です。その場合はサンプルを-80℃で保存します。
- 細胞/組織を溶解し、クロマチン画分を回収する。
- クロマチンを断片化する(超音波処理)。
この段階で操作を中断することが可能です。その場合はサンプルを-80℃で保存します。
- DNAと結合するタンパク質に特異的な抗体を使用して免疫沈降(IP)/プルダウンを実施する。
- タンパク質‐DNA複合体の脱クロスリンクを実施し、タンパク質とDNAを分離させる。
- DNAフラグメントを精製しクリーンアップする。
- DNA解析を実施する(PCR、qPCR、マイクロアレイ、シーケンス解析)。
ChIP実験を成功させるポイント
ポイント1:常に細胞/組織を氷上で取り扱います
- 温度はサンプルに重大な影響を及ぼします。細胞溶解は4℃で実施します。
- サンプルは氷冷し、バッファーも氷冷したものを使用します。
ポイント2:不十分/過剰な架橋(クロスリンク)
- 架橋時間とホルムアルデヒド濃度の両方が重要となります。
- 注記:パラホルムアルデヒド溶液を使用する場合は、新たに調製されたものであることを確認してください(終濃度が1%~5%になるよう添加します)。
- 架橋時間も重要なポイントで、クロスリンクが不十分になったり過剰になったりします。
| 不十分なクロスリンク | 過剰なクロスリンク |
| タンパク質‐DNA複合体が脱会合し、複合体の回収量が低下します。 | 抗体が結合するための重要なエピトープ領域がマスクされ、細胞溶解の効率が下がり、クロマチンのせん断が妨げられると共に、タンパク質‐DNA複合体が脱クロスリンクできなくなります。 |
ポイント3:クロマチンせん断および超音波処理
- 組織懸濁液中にヌクレオソームの長鎖フラグメントが存在しないようにします。
- 先端をカットしたピペットチップを使用して、組織を十分にホモジナイズします。
- 超音波照射装置のプローブがチューブの壁に接触しないようにセットします。 超音波処理の回数を増やすと、よりクロマチンはせん断されます。ただし、1回の照射時間(または出力数)は増やさないでください。サンプルが過剰に熱くなり、抗原性が喪失するおそれがあります。
- 超音波照射装置を操作する際は氷を使用し、サンプルが過剰に熱くなるのを防ぎます。
- クロマチンの超音波処理によって、サイズが500 bp未満になることは避けてください(アガロースゲル電気泳動を実施し、分子量を確認します)。
- 注記:細胞の種類が異なると、DNA断片化に最適な条件も異なる可能性があります。適切な長さのDNAフラグメントが得られるよう超音波処理時間を最適化してください。
- 定量分析用のコントロールサンプル(inputサンプル)を準備してください。コントロールサンプル中のDNA量に対して、免疫沈降後サンプルのDNA量を最終的に比較します。
ポイント4:免疫沈降(IP)
- 希釈およびプレクリア処理したChIP画分をIP画分とみなして、この画分を目的タンパク質に対する抗体またはコントロール抗体と共に免疫沈降に使用します。
- 1回のIPに約25µgのDNAを使用します(機能的なIPのためには最低でも1x106以上の細胞数が必要であり、ChIPの場合の目安となる細胞数は1x107 cells程度です)。
- DNA 25µgあたり、1~10µgの抗体を使用します。
- 抗体の中には、細胞ライセートとのインキュベーションを室温・短時間で実施しても問題ない抗体もありますが、一般的には4℃でovernightインキュベーションして、シグナル強度や特異性を高めます。
- 再現性のある実験データを得るために、すべてのサンプルに等量のプロテインA/Gアガロースビーズ、または磁気ビーズを添加します。ビーズは添加前にピペッティングを行い十分に懸濁します。
| 抗体の選択について | |
| 抗体の選択 | ChIPに使用される抗体は、タンパク質‐DNA複合体上に露出しているエピトープを認識できる必要があります。IHC(免疫組織化学)、WB(ウェスタンブロット)、IF(免疫蛍光染色)に使用できる抗体でも、ChIPには使用できない場合があります。 |
| 抗体の特異性 | 抗体が特異的にタンパク質‐DNA複合体と結合できているか否かを確認するために、IP後にWBやELISAにより検証を実施します。 |
| 抗体の濃度 | 極端に抗体濃度が高いと、PCRバックグラウンドが高くなる原因になる場合があります。 抗体濃度が低いと、タンパク質‐DNA複合体の回収率が低下し、PCRを実施するために必要な量のDNAを回収できなくなります。 |
- ビーズの準備:プロテインAとプロテインG結合ビーズを使用する場合は、等量を混和します
- ビーズは、RIPAバッファー中で複数回洗浄します。
- ビーズをブロッキングし、非特異的結合を防ぎます。
- ビーズをIP用のサンプルに添加し、4℃でovernightインキュベーションします。
| ビーズの選択について | |
| 磁気ビーズ | 磁力を利用して簡便に分離・回収可能で、チューブ内での視認性が良好な、サンプルロスの少ないビーズです。 |
| アガロースビーズ | 多孔質という特性から表面積が広く、高い結合能を示します。 |
| プロテインAビーズ | ウサギポリクローナル抗体に非常に高い親和性を示します。 |
| プロテインGビーズ | 様々な抗体に非常に高い親和性を示します。 |
| プロテインA/Gビーズ | プロテインAとプロテインGの両方をコートしたビーズで、様々な抗体と高い親和性で結合します。 |
- 各実験に、陽性コントロールおよび陰性コントロールを設定します。
- ゲノム領域と相互作用することが判明しているタンパク質、およびゲノム領域とは反応しないことが判明しているタンパク質を利用して、それぞれ陽性コントロール、陰性コントロールとしてIPを実施し、結果を判定します。
| ChIPのコントロールについて | |
| 抗体コントロール | 一般的に、陽性コントロールとしてヒストンH3抗体が使用されています。陰性コントロールには、GFP抗体等のクロマチンに存在しないエピトープを認識する抗体が使用されています。 |
| 陰性コントロール | 抗体を使用せずビーズのみで反応を行うか、ビーズと免疫グロブリン(Ig)コントロール抗体を反応させ、測定系のバックグラウンドを確認します。 |
| 遺伝子座コントロール | ChIPで対象とするタンパク質に結合することが判明している遺伝子、および結合しないことが判明している遺伝子を、それぞれ陽性コントロール、陰性コントロールとします。各遺伝子を検出するプライマーを用意し、免疫沈降後に得られるサンプルを鋳型としてRT-PCRを実施し、発現の有無や発現量を確認します。 |
| PCRコントロール | 鋳型を添加しないコントロールサンプルを調製してPCRを実施し、コンタミネーションの有無を確認します。 |
ビーズに関する注意点
- 常に、ピペッティングする前にボルテックスを実施し、ビーズを完全に再懸濁します。
- 常に4°Cで保管し、決してビーズが乾かないようにします。
- 抗体のサブクラスがプロテインA/Gと適合性があることを確認します。
抗体に関する注意点
- 目的の抗体がChIPに使用されている実績があるかどうかを確認します(抗体の特異性は、IP後にWBを実施することで確認できます)。
ポイント5:脱クロスリンク
- 一般的に、95℃で15分間インキュベーションすることで、十分に脱クロスリンクできます(タンパク質‐DNA複合体がタンパク質とDNAに分離します)。
- 一部のサンプルは、65°Cで4時間またはovernight (16時間以上)のインキュベーションを実施し、場合によってはその後プロテイナーゼK処理を併用する必要があります。
ポイント6:DNAの溶出と精製
- 異なる洗浄バッファー(例:塩濃度が低いバッファーと高いバッファー、LiClバッファーとTEバッファー)を使用します。
- 市販の精製カラムを使用する際は、洗浄ステップ後にカラムが完全に乾いていることを確認してください。洗浄バッファーが残っていると溶出が阻害されます。
- 必ず溶出バッファーをカラム内のシリカ膜上に直接添加し、1分間以上なじませてからDNAの溶出を実施します。
- コンタミネーションを防止するために、実験ごとにストック溶液から新たに試薬を調製します。
- PCR産物のシグナルが微弱である、または増幅しない場合は、ヌクレオソームを形成しない領域にプライマーを設計してしまっているため、DNAを増幅できていない可能性があります。
- 注記:プライマー配列とその組み合わせ、プライマー濃度、場合によってはPCR条件等を変更し、「ゲノムDNA」を鋳型にして検討を実施します。鋳型としてStandard DNAまたはinput DNAを使用することは、適切な増幅産物を得るためのプライマーを選択することに役立ちます。
- 鋳型DNAが適切な濃度となるよう調節し、PCRの条件を最適化します。必要に応じて鋳型DNAの添加量を増やすか、増幅サイクルの回数を増やします。
- 各操作で設定した陽性コントロールサンプルの増幅産物が認められない場合は、IPによってクロマチンが十分に回収できていたか、あるいは十分な量のターゲットを捕捉できる濃度の抗体を添加できていたか確認します。
- クロマチンをプロテインA/Gビーズから完全に溶出してください。バッファーに懸濁したビーズを65℃でミキシングすると(~10分間)、良好にクロマチンを溶出することができます。
覚えておくべき重要なポイント
- すべての操作は氷上で実施し、試薬を低温に保ってください。
- サンプルごとにクロマチンのせん断条件を最適化してください。
- 使用する抗体がChIPで使用できるか検証されているとは限らないため、情報を収集して実際に検証を実施してください。
- 信頼できる結果を得るために、陽性コントロールと陰性コントロールを設定してください。
- 脱クロスリンクの条件を最適化してください。
- 得られたDNAを解析する前に、精製を実施し濃度を測定してください。
New chat
Able™
正在加载,请稍候...